

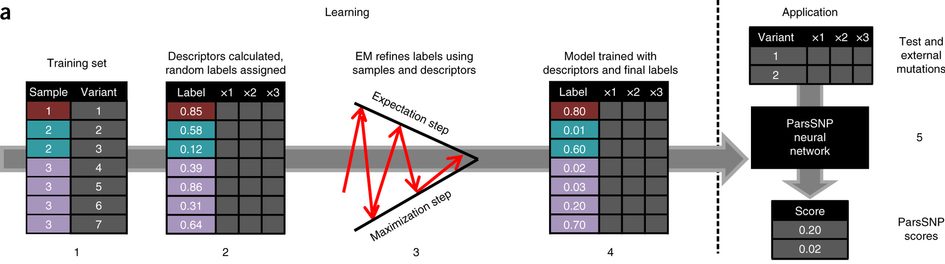
This study uses��an algorithm first developed�in our group for modeling�metabolism from ambiguous data. Often we know the region of a molecule that is metabolized, but not the specific atom. A clever trick using the EM algorithm lets us train a neural network, even without�knowing the right targets.
An analogous problem arises in cancer genomics. We know from both experimental studies and the neutral theory of evolution,1 that only a few mutations cause cancer�(the drivers), but most are neutral with no functional role (the passengers). �This leaves us with a fundamentally important question:
When we sequence a new cancer’s genome, how do we determine which mutations�are the drivers?
Prior methods used�training data where mutations were labeled as drivers or passengers. These methods, however, are only as good as their training data. In�our new�approach works better than all prior methods, even though it ignores�known drivers and passengers when training. �An�unsupervised method�(ParsSNP), guided by�parsimony, works better than approaches guided by our current knowledge of drivers.
ParsSNP�starts from a “parsimony” assumption, rooted in the neutral theory of evolution; we assume driver mutations are rare and equally distributed among patients.�Using this parsimony assumption, we can�now train a model to identify the drivers, without knowing ahead of times which ones are which.
Not surprisingly, the model relies directly evolutionary theory to make its predictions. Several evolutionary�features�(including, for example, Ks/Ka ratio) are used to determine which mutations are driver. So evolutionary theory helps solve a key scientific problem in cancer in two ways: both inspiring �a new computational algorithm and the right way to mathematically describe mutations.
We hope this study will open whole new ways of studying genomic data because it does not require labeled training data.
Runjun Kumar (pictured above), a�MD PhD student in Dr. Ron Bose’s group here at WUSTL. Runjun did a massive amount of work to collate the required data, train the models, and test it against other methods.
Unsupervised cancer driver detection with parsimony-guided learning.
Runjun D. Kumar, S. Joshua Swamidass*, and Ron Bose*�
*Co-corresponding authors
Abstract:
Methods are needed to reliably prioritize biologically active driver mutations over inactive passengers in high-throughput cancer sequencing datasets. We present ParsSNP, an unsupervised functional impact predictor that is guided by parsimony. ParsSNP uses an expectation-maximization framework to find mutations that explain tumor incidence broadly, without using pre-defined training labels that can introduce bias. We compare ParsSNP to five existing tools (CanDrA, CHASM, FATHMM Cancer, TransFIC, Condel) across five distinct benchmarks. ParsSNP outperformed existing tools in 24 out of 25 comparisons. To investigate the real-world benefit of these improvements, ParsSNP was applied to an independent dataset of thirty patients with diffuse-type gastric cancer. It identified many known and likely driver mutations that other methods did not detect, including truncation mutations in known tumor suppressors and the recurrent driver RHOA Y42C. In conclusion, ParsSNP uses an innovative, parsimony-based approach to prioritize cancer drivers and provides dramatic improvements over existing methods.
http://www.nature.com/ng/journal/vaop/ncurrent/full/ng.3658.html
You must log in to post a comment.