
Article available at:�https://pubs.acs.org/doi/full/10.1021/acscentsci.7b00405.
Matthew K. Matlock, Na Le Dang, and S. Joshua Swamidass
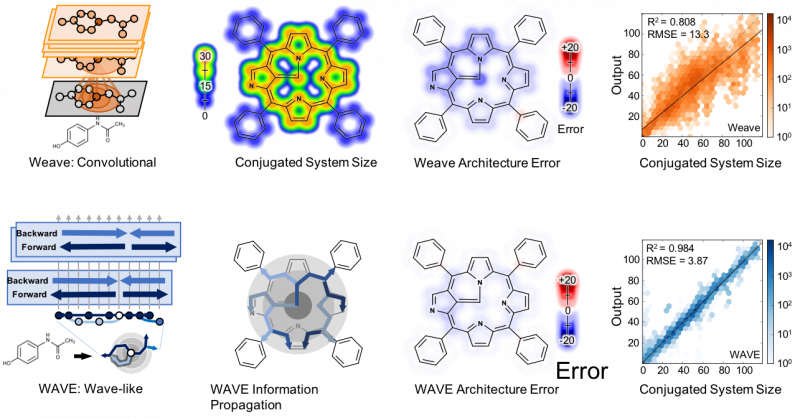
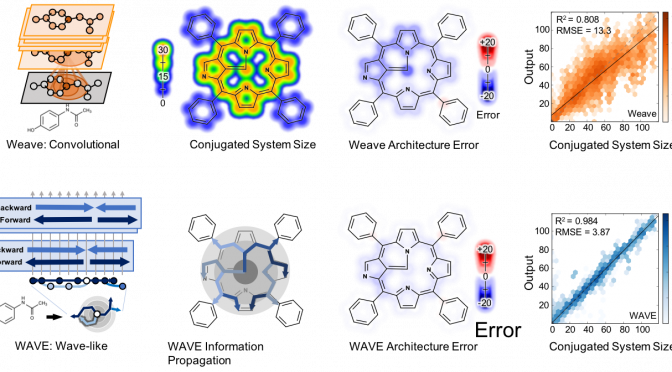
Abstract: A collection of new approaches to building and training neural networks, collectively referred to as deep learning, are attracting attention in theoretical chemistry. Several groups aim to replace computationally expensive ab initio quantum mechanics calculations with learned estimators. This raises questions about the representability of complex quantum chemical systems with neural networks. Can local-variable models efficiently approximate nonlocal quantum chemical features? Here, we find that convolutional architectures, those that only aggregate information locally, cannot efficiently represent aromaticity and conjugation in large systems. They cannot represent long-range nonlocality known to be important in quantum chemistry. This study uses aromatic and conjugated systems computed from molecule graphs, though reproducing quantum simulations is the ultimate goal. This task, by definition, is both computable and known to be important to chemistry. The failure of convolutional architectures on this focused task calls into question their use in modeling quantum mechanics. To remedy this heretofore unrecognized deficiency, we introduce a new architecture that propagates information back and forth in waves of nonlinear computation. This architecture is still a local-variable model, and it is both computationally and representationally efficient, processing molecules in sublinear time with far fewer parameters than convolutional networks. Wave-like propagation models aromatic and conjugated systems with high accuracy, and even models the impact of small structural changes on large molecules. This new architecture demonstrates that some nonlocal features of quantum chemistry can be efficiently represented in local variable models.