
Modeling Reactivity to Biological Macromolecules with a Deep Multitask Network
Hughes, T. B., Dang, N. L., Miller, G. P., and Swamidass, S. J. (2016). �ACS Central Science 2(8), 529�537.
Abstract:
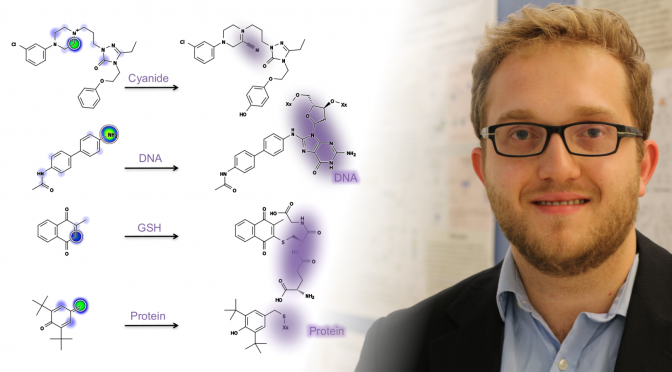
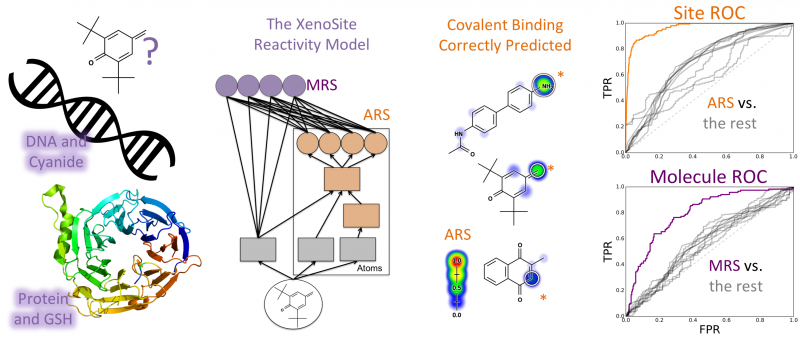
Most small-molecule drug candidates fail before entering the market, frequently because of unexpected toxicity. Often, toxicity is detected only late in drug development, because many types of toxicities, especially idiosyncratic adverse drug reactions (IADRs), are particularly hard to predict and detect. Moreover, drug-induced liver injury (DILI) is the most frequent reason drugs are withdrawn from the market and causes 50% of acute liver failure cases in the United States. A common mechanism often underlies many types of drug toxicities, including both DILI and IADRs. Drugs are bioactivated by drug-metabolizing enzymes into reactive metabolites, which then conjugate to sites in proteins or DNA to form adducts. DNA adducts are often mutagenic and may alter the reading and copying of genes and their regulatory elements, causing gene dysregulation and even triggering cancer. Similarly, protein adducts can disrupt their normal biological functions and induce harmful immune responses. Unfortunately, reactive metabolites are not reliably detected by experiments, and it is also expensive to test drug candidates for potential to form DNA or protein adducts during the early stages of drug development. In contrast, computational methods have the potential to quickly screen for covalent binding potential, thereby flagging problematic molecules and reducing the total number of necessary experiments. Here, we train a deep convolution neural network�the XenoSite reactivity model�using literature data to accurately predict both sites and probability of reactivity for molecules with glutathione, cyanide, protein, and DNA. On the site level, cross-validated predictions had area under the curve (AUC) performances of 89.8% for DNA and 94.4% for protein. Furthermore, the model separated molecules electrophilically reactive with DNA and protein from nonreactive molecules with cross-validated AUC performances of 78.7% and 79.8%, respectively. On both the site- and molecule-level, the model�s performances significantly outperformed reactivity indices derived from quantum simulations that are reported in the literature. Moreover, we developed and applied a selectivity score to assess preferential reactions with the macromolecules as opposed to the common screening traps. For the entire data set of 2803 molecules, this approach yielded totals of 257 (9.2%) and 227 (8.1%) molecules predicted to be reactive only with DNA and protein, respectively, and hence those that would be missed by standard reactivity screening experiments. Site of reactivity data is an underutilized resource that can be used to not only predict if molecules are reactive, but also show where they might be modified to reduce toxicity while retaining efficacy. The XenoSite reactivity model is available at https://swami.wustl.edu/xenosite/p/reactivity.
Publication (open access):
You must log in to post a comment.